如果你也在 怎样代写信息论information theory 这个学科遇到相关的难题,请随时右上角联系我们的24/7代写客服。信息论information theory的一个关键衡量标准是熵。熵量化了随机变量的值或随机过程的结果中所涉及的不确定性的数量。例如,确定一个公平的抛硬币的结果(有两个同样可能的结果)比确定一个掷骰子的结果(有六个同样可能的结果)提供的信息要少(熵值较低)。
信息论information theory基本课题的应用包括源编码/数据压缩(如ZIP文件),以及信道编码/错误检测和纠正(如DSL)。它的影响对于旅行者号深空任务的成功、光盘的发明、移动电话的可行性和互联网的发展都至关重要。该理论在其他领域也有应用,包括统计推理、密码学、神经生物学、感知、语言学、分子代码的进化和功能(生物信息学)、热物理、分子动力学、量子计算、黑洞、信息检索、情报收集、剽窃检测、模式识别、异常检测甚至艺术创作。
my-assignmentexpert™信息论information theory代写,免费提交作业要求, 满意后付款,成绩80\%以下全额退款,安全省心无顾虑。专业硕 博写手团队,所有订单可靠准时,保证 100% 原创。my-assignmentexpert, 最高质量的信息论information theory作业代写,服务覆盖北美、欧洲、澳洲等 国家。 在代写价格方面,考虑到同学们的经济条件,在保障代写质量的前提下,我们为客户提供最合理的价格。 由于统计Statistics作业种类很多,同时其中的大部分作业在字数上都没有具体要求,因此信息论information theory作业代写的价格不固定。通常在经济学专家查看完作业要求之后会给出报价。作业难度和截止日期对价格也有很大的影响。
想知道您作业确定的价格吗? 免费下单以相关学科的专家能了解具体的要求之后在1-3个小时就提出价格。专家的 报价比上列的价格能便宜好几倍。
my-assignmentexpert™ 为您的留学生涯保驾护航 在澳洲代写方面已经树立了自己的口碑, 保证靠谱, 高质且原创的澳洲代写服务。我们的专家在信息论information theory代写方面经验极为丰富,各种信息论information theory相关的作业也就用不着 说。
我们提供的信息论information theory及其相关学科的代写,服务范围广, 其中包括但不限于:
数学代写|信息论代写Information Theory代考|The structure of DNA
Until 1944, most scientists had guessed that the genetic message was carried by the proteins of the chromosome. In 1944, however, O.T. Avery and his co-workers at the laboratory of the Rockefeller Institute in New York performed a critical experiment, which proved that the material which carries genetic information is not protein, but deoxyribonucleic acid (DNA) a giant chainlike molecule which had been isolated from cell nuclei by the Swiss chemist, Friedrich Miescher.
Avery had been studying two different strains of pneumococci, the bacteria which cause pneumonia. One of these strains, the S-type, had a smooth coat, while the other strain, the R-type, lacked an enzyme needed for the manufacture of a smooth carbohydrate coat. Hence, R-type pneumococci had a rough appearance under the microscope. Avery and his co-workers were able to show that an extract from heat-killed S-type pneumococci could convert the living R-type species permanently into Stype; and they also showed that this extract consisted of pure DNA.
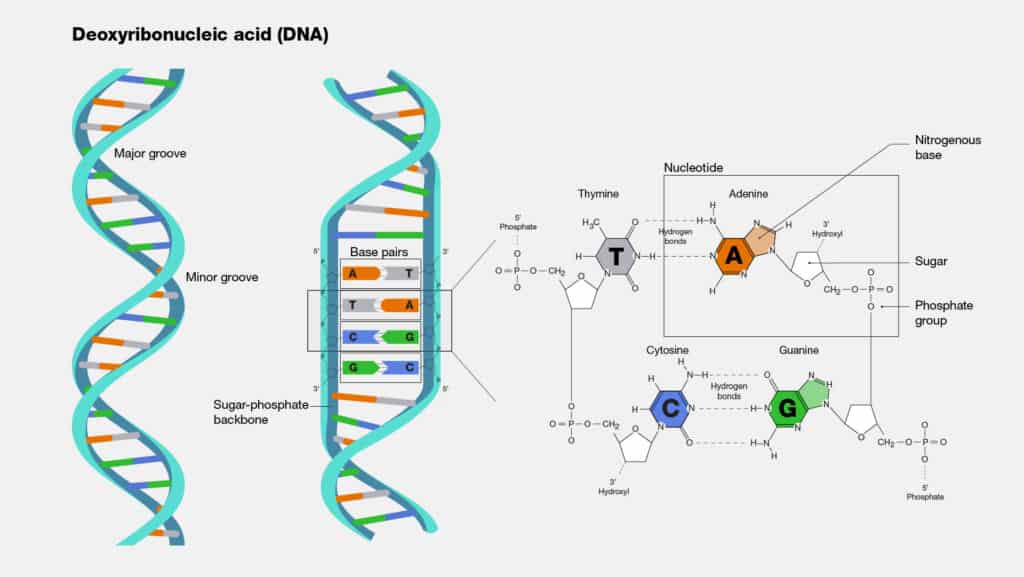
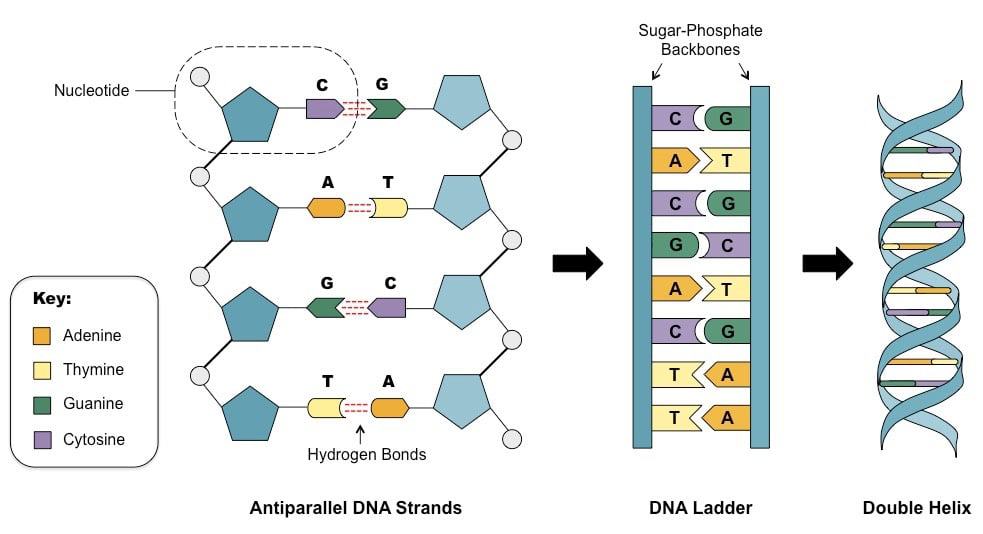
In 1947, the Austrian-American biochemist, Erwin Chargaff, began to study the long, chain-like DNA molecules. It had already been shown by Levine and Todd that chains of DNA are built up of four bases: adenine (A), thymine $(\mathrm{T})$, guanine $(\mathrm{G})$ and cytosine $(\mathrm{C})$, held together by a sugarphosphate backbone. Chargaff discovered that in DNA from the nuclei of living cells, the amount of $\mathrm{A}$ always equals the amount of $\mathrm{T}$; and the amount of $\mathrm{G}$ always equals the amount of $\mathrm{C}$.
When Chargaff made this discovery, neither he nor anyone else understood its meaning. However, in 1953, the mystery was completely solved by Rosalind Franklin and Maurice Wilkins at Kings College, London, together with James Watson and Francis Crick at Cambridge University. By means of X-ray diffraction techniques, Wilkins and Franklin obtained crystallographic information about the structure of DNA. Using this information, together with Linus Pauling’s model-building methods, Crick and Watson proposed a detailed structure for the giant DNA molecule.
数学代写|信息论代写Information Theory代考|Protein structure
In England, J.D. Bernal and Dorothy Crowfoot Hodgkin pioneered the application of X-ray diffraction methods to the study of complex biological molecules. In 1949, Hodgkin determined the structure of penicillin; and in 1955, she followed this with the structure of vitamin B12. In 1960, Max Perutz and John C. Kendrew obtained the structures of the blood proteins myoglobin and hemoglobin. This was an impressive achievement for the Cambridge crystallographers, since the hemoglobin molecule contains roughly 12,000 atoms.
The structure obtained by Perutz and Kendrew showed that hemoglobin is a long chain of amino acids, folded into a globular shape, like a small, crumpled ball of yarn. They found that the amino acids with an affinity for water were on the outside of the globular molecule; while the amino acids for which contact with water was energetically unfavorable were hidden on the inside. Perutz and Kendrew deduced that the conformation of the protein – the way in which the chain of amino acids folded into a 3dimensional structure – was determined by the sequence of amino acids in the chain.
In 1966, D.C. Phillips and his co-workers at the Royal Institution in London found the crystallographic structure of the enzyme lysozyme an egg-white protein which breaks down the cell walls of certain bacteria. Again, the structure showed a long chain of amino acids, folded into a roughly globular shape. The amino acids with hydrophilic groups were on the outside, in contact with water, while those with hydrophobic groups were on the inside. The structure of lysozyme exhibited clearly an active site, where sugar molecules of bacterial cell walls were drawn into a mouthlike opening and stressed by electrostatic forces, so that bonds between the sugars could easily be broken.
Meanwhile, at Cambridge University, Frederick Sanger developed methods for finding the exact sequence of amino acids in a protein chain. In 1945 , he discovered a compound (2,4-dinitrofluorobenzene) which attaches itself preferentially to one end of a chain of amino acids. Sanger then broke down the chain into individual amino acids, and determined which of them was connected to his reagent. By applying this procedure many times to fragments of larger chains, Sanger was able to deduce the sequence of amino acids in complex proteins. In 1953, he published the sequence of insulin. This led, in 1964 , to the synthesis of insulin.
The biological role and structure of proteins which began to emerge was as follows: A mammalian cell produces roughly 10,000 different proteins. All enzymes are proteins; and the majority of proteins are enzymes – that is, they catalyze reactions involving other biological molecules. All proteins are built from chainlike polymers, whose monomeric sub-units are the following twenty amino acids: glycine, aniline, valine, isoleucine, leucine, serine, threonine, proline, aspartic acid, glutamic acid, lysine, arginine, asparagine, glutamine, cysteine, methionine, tryptophan, phenylalanine, tyrosine and histidine. These individual amino acid monomers may be connected together into a polymer (called a polypeptide) in any order – hence the great number of possibilities. In such a polypeptide, the backbone is a chain of carbon and nitrogen atoms showing the pattern …-C-C-N-C-C-NC-C-N-… and so on. The -C-C-N- repeating unit is common to all amino acids. Their individuality is derived from differences in the side groups which are attached to the universal -C-C-N- group.
信息论代写
数学代写|信息论代写INFORMATION THEORY代考|THE STRUCTURE OF DNA
直到 1944 年,大多数科学家都猜测遗传信息是由染色体的蛋白质携带的。然而,1944年,纽约洛克菲勒研究所实验室的OT Avery和他的同事进行 了一项关键实验,证明携带遗传信息的物质不是蛋白质,而是脱氧核糖核酸 $D N A$ 瑞士化学家 Friedrich Miescher 从细胞核中分离出来的巨大链状 分子。
艾弗里一直在研究两种不同的肺谈球菌菌株,即引起肺炎的细䒩。其中一种菌株, $\mathrm{S}$ 型,具有光滑的外壳,而另一种蒠株, $\mathrm{R}$ 型,缺乏制造光滑的 碳水化合物外壳所需的酶。因此,R型肺炎球菌在显微鏡下具有粗粘的外观。Avery 和他的同事能够证明,从热灭活的 $\mathrm{S}$ 型肺炎球菌中提取的提取 物可以将活的 $\mathrm{R}$ 型肺炎球菌永久性地转化为 $\mathrm{S}$ 型;他们还表明,这种提取物由纯 DNA 组成。
1947 年,美籍奧地利生物化学家 Erwin Chargaff 开始研究长链状 DNA 分子。Levine和 Todd 已经证明,DNA 链由四个碱基组成:腺嘌呤 $A$, 胸腺嘧啶 $(T)$, 鸟嘌呤 $(G)$ 和胞嘧啶 $(\mathrm{C})$, 由糖磷酸主链结合在一起。Chargaff 发现,在活细胞核的 DNA 中,A总是等于T; 和数量 $G$ 总是等于C.
当 Chargaff 做出这一发现时,他和其他任何人都不明白其中的含义。然而,在 1953 年,伦敦国王学院的 Rosalind Franklin 和 Maurice Wilkins 以及 剑桥大学的 James Watson 和 Francis Crick 彻底解开了这个谜团。借助 X射线衍射技术,威尔金斯和富兰克林获得了有关 DNA 结构的晶体学信息。 利用这些信息,连同莱纳斯鲍林的模型构建方法,克里克和沃森提出了巨型 DNA 分子的详细结构。
数学代写|信息论代写INFORMATION THEORY代考|PROTEIN STRUCTURE
在英国,JD Bernal 和 Dorothy Crowfoot Hodgkin 率先将 X 射线衍射方法应用于复杂生物分子的研究。1949年,霍奇金确定了青霉素的结构;1955 年,她沿用了维生素 B12 的结构。1960 年,Max Perutz 和 John C. Kendrew 获得了血液蛋白肌红蛋白和血红蛋白的结构。对于剑桥大学的晶体学家来说,这是一项了不起的成就,因为血红蛋白分子包含大约 12,000 个原子。
Perutz 和 Kendrew 获得的结构表明,血红蛋白是一种长链氨基酸,折叠成球状,就像一个皱巴巴的小纱线球。他们发现,对水有亲和力的氨基酸位于球状分子的外侧;而在能量上不利于与水接触的氨基酸则隐藏在内部。Perutz 和 Kendrew 推断蛋白质的构象——氨基酸链折叠成三维结构的方式——是由链中的氨基酸序列决定的。
1966 年,DC Phillips 和他在伦敦皇家研究所的同事发现了溶菌酶的晶体结构,溶菌酶是一种蛋清蛋白,可以分解某些细菌的细胞壁。同样,该结构显示了一条长链氨基酸,折叠成大致球形。带有亲水基团的氨基酸在外面,与水接触,而带有疏水基团的氨基酸在里面。溶菌酶的结构清楚地展示了一个活性位点,细菌细胞壁的糖分子被吸引到嘴状开口中并受到静电力的压力,因此糖之间的键很容易被破坏。
与此同时,在剑桥大学,弗雷德里克.桑格 (Frederick Sanger) 开发了寻找蛋白质链中氨基酸准确序列的方法。1945年,他发现了一种化合物 2, 4-dinitrofluorobenzene 它优先附着在氨基酸链的一端。桑格随后将这条链分解成单个氨基酸,并确定其中哪些与他的试剂有关。通过将此 过程多次应用于较大链的片段,桑格能够推断出复杂蛋白质中的氨基酸序列。1953年,他发表了胰岛素序列。这导致了1964年胰岛素的合成。
开始出现的蛋白质的生物学作用和结构如下:哺乳动物细胞产生大约 10,000 种不同的蛋白质。所有的酶都是蛋白质;大多数蛋白质都是酶一一也就 是说,它们催化涉及其他生物分子的反应。所有蛋白质均由链状聚合物构成,其单体亚基为以下二十种氨基酸:甘氞酸、苯胺、缬氨酸、异亮氨 酸、亮氨酸、丝氨酸、苏氨酸、脯氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸、天冬酰胺、谷氨酰胺、半胱氨酸、甲硫氨酸、色氨酸、苯丙氨 酸、酪氨酸和组氨酸。这些单独的氨基酸单体可以连接在一起形成聚合物calledapolypeptide以任何顺序- 因此有大量的可能性。在这样的多肽 中,主链是碳原子和氮原子链,显示出…-CCNCC-NC-CN-..等模式。-CCN-重复单元是所有氨基酸共有的。它们的个性源于与通用-CCN-基团相连的 侧基的差异。

数学代写|信息论代写Information Theory代考 请认准UprivateTA™. UprivateTA™为您的留学生涯保驾护航。
微观经济学代写
微观经济学是主流经济学的一个分支,研究个人和企业在做出有关稀缺资源分配的决策时的行为以及这些个人和企业之间的相互作用。my-assignmentexpert™ 为您的留学生涯保驾护航 在数学Mathematics作业代写方面已经树立了自己的口碑, 保证靠谱, 高质且原创的数学Mathematics代写服务。我们的专家在图论代写Graph Theory代写方面经验极为丰富,各种图论代写Graph Theory相关的作业也就用不着 说。
线性代数代写
线性代数是数学的一个分支,涉及线性方程,如:线性图,如:以及它们在向量空间和通过矩阵的表示。线性代数是几乎所有数学领域的核心。
博弈论代写
现代博弈论始于约翰-冯-诺伊曼(John von Neumann)提出的两人零和博弈中的混合策略均衡的观点及其证明。冯-诺依曼的原始证明使用了关于连续映射到紧凑凸集的布劳威尔定点定理,这成为博弈论和数学经济学的标准方法。在他的论文之后,1944年,他与奥斯卡-莫根斯特恩(Oskar Morgenstern)共同撰写了《游戏和经济行为理论》一书,该书考虑了几个参与者的合作游戏。这本书的第二版提供了预期效用的公理理论,使数理统计学家和经济学家能够处理不确定性下的决策。
微积分代写
微积分,最初被称为无穷小微积分或 “无穷小的微积分”,是对连续变化的数学研究,就像几何学是对形状的研究,而代数是对算术运算的概括研究一样。
它有两个主要分支,微分和积分;微分涉及瞬时变化率和曲线的斜率,而积分涉及数量的累积,以及曲线下或曲线之间的面积。这两个分支通过微积分的基本定理相互联系,它们利用了无限序列和无限级数收敛到一个明确定义的极限的基本概念 。
计量经济学代写
什么是计量经济学?
计量经济学是统计学和数学模型的定量应用,使用数据来发展理论或测试经济学中的现有假设,并根据历史数据预测未来趋势。它对现实世界的数据进行统计试验,然后将结果与被测试的理论进行比较和对比。
根据你是对测试现有理论感兴趣,还是对利用现有数据在这些观察的基础上提出新的假设感兴趣,计量经济学可以细分为两大类:理论和应用。那些经常从事这种实践的人通常被称为计量经济学家。
Matlab代写
MATLAB 是一种用于技术计算的高性能语言。它将计算、可视化和编程集成在一个易于使用的环境中,其中问题和解决方案以熟悉的数学符号表示。典型用途包括:数学和计算算法开发建模、仿真和原型制作数据分析、探索和可视化科学和工程图形应用程序开发,包括图形用户界面构建MATLAB 是一个交互式系统,其基本数据元素是一个不需要维度的数组。这使您可以解决许多技术计算问题,尤其是那些具有矩阵和向量公式的问题,而只需用 C 或 Fortran 等标量非交互式语言编写程序所需的时间的一小部分。MATLAB 名称代表矩阵实验室。MATLAB 最初的编写目的是提供对由 LINPACK 和 EISPACK 项目开发的矩阵软件的轻松访问,这两个项目共同代表了矩阵计算软件的最新技术。MATLAB 经过多年的发展,得到了许多用户的投入。在大学环境中,它是数学、工程和科学入门和高级课程的标准教学工具。在工业领域,MATLAB 是高效研究、开发和分析的首选工具。MATLAB 具有一系列称为工具箱的特定于应用程序的解决方案。对于大多数 MATLAB 用户来说非常重要,工具箱允许您学习和应用专业技术。工具箱是 MATLAB 函数(M 文件)的综合集合,可扩展 MATLAB 环境以解决特定类别的问题。可用工具箱的领域包括信号处理、控制系统、神经网络、模糊逻辑、小波、仿真等。